Project
Novel Materials by Design: The Treasure Map Approach
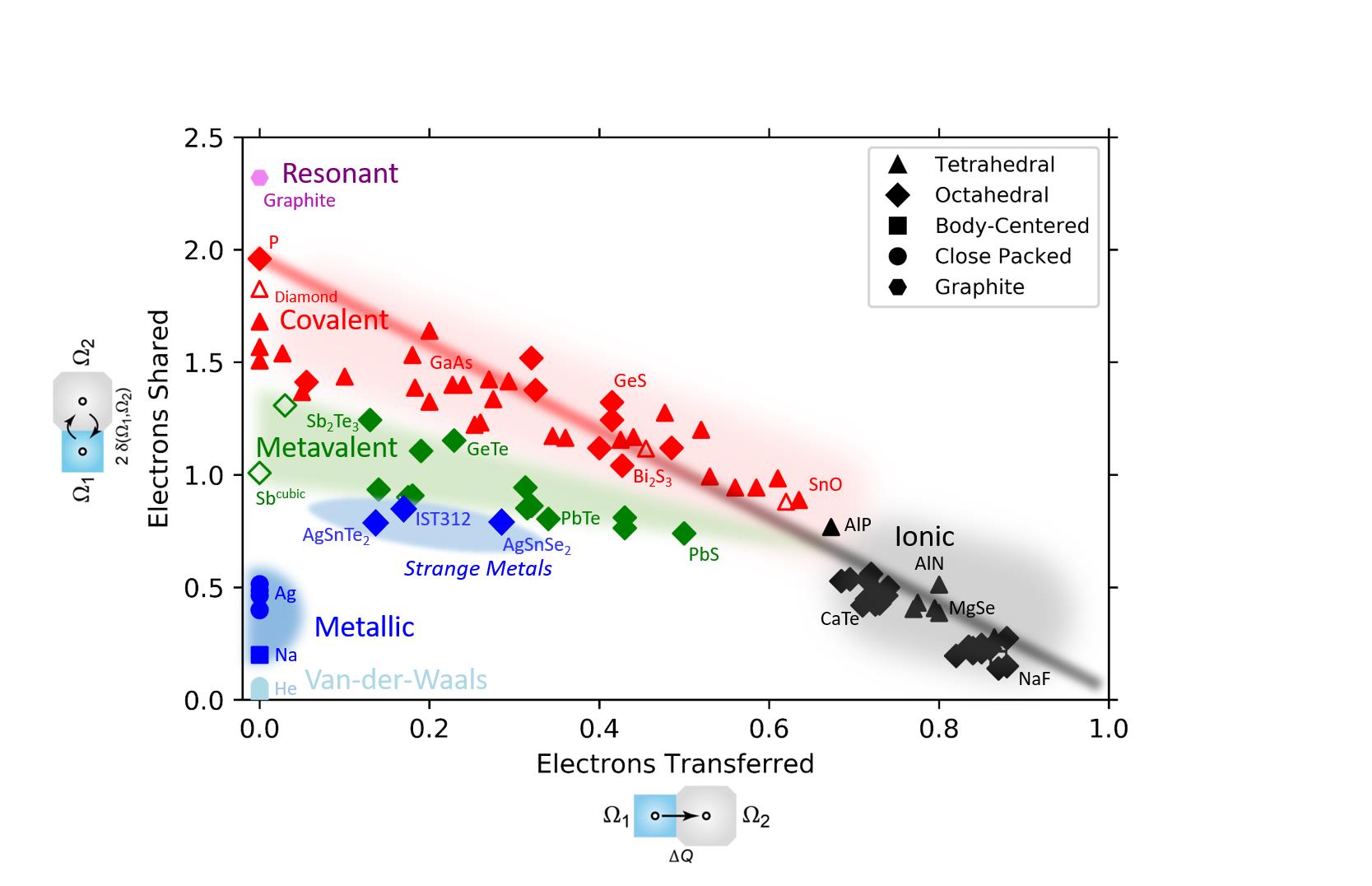 Figure 1: Map based on quantum-chemical calculations performed within this project. Upon determination of the sharing and transfer of electrons between adjacent atoms and the corresponding material properties we can distinguish three fundamental bonding mechanisms (metallic, covalent and ionic bonding) and can identify a novel fourth bonding mechanism, i.e. ‘metavalent bonding’. Furthermore, the so-called group of “Strange Metals” (which still belong to the general group of metals) are located right below the metavalent region.
Figure 1: Map based on quantum-chemical calculations performed within this project. Upon determination of the sharing and transfer of electrons between adjacent atoms and the corresponding material properties we can distinguish three fundamental bonding mechanisms (metallic, covalent and ionic bonding) and can identify a novel fourth bonding mechanism, i.e. ‘metavalent bonding’. Furthermore, the so-called group of “Strange Metals” (which still belong to the general group of metals) are located right below the metavalent region. Employing density functional theory (DFT) calculations and the Quantum Theory of Atoms in Molecules (QTAIM), we are able to extract two quantities for atoms in a molecule, that let us distinguish different bonding mechanisms. These two quantities are the electrons shared (ES) and electrons transferred (ET). A map for various materials can be seen in Fig. 1. The three main bonding mechanisms visible in the map are covalent, ionic and metallic bonding. There is also a fourth, unconventional bonding mechanism, which we refer to as ´metavalent bonding´. The map can help to explain and predict property trends as shown on the following website: https://materials-map.rwth-aachen.de/.
Project Details
Project term
January 1, 2021–December 31, 2021
Affiliations
RWTH Aachen University
University of Liège
Institute
Institute of Physics
Project Manager
Principal Investigator
Methods
Part of this project is the exploration of novel photovoltaic materials, employing the excellent predictive properties of the aforementioned map. As an example, there is a direct link between the number of electrons transferred and the band gap of a material in the metavalent region.
Results
We have already calculated ES and ET for various double halide perovskites (structure AB’B’’X6, where X denotes a halogen atom), which seem to be a promising alternative to halide perovskites (structure ABX3), which are being used in photovoltaics right now. Furthermore, other properties, such as the frequency dependent dielectric function, the band structure and effective masses were determined via DFT calculations. In addition to that, we calculated ES and ET for other known photovoltaic materials, such as III-V-semiconductors and Silicon. In this early stage of the project we are still in the process of selecting the right properties to study photovoltaic material trends in the map. Also, we are optimizing the obtained quantities for accuracy employing different functionals (e.g. HSE06).
Discussion
However, we already published a couple of papers containing results of this project, see Publications.
Additional Project Information
DFG classification: 307-02 Theoretical Condensed Matter Physics
Software: QuantumEspresso, ABINIT, VASP, DGRID, Critic2, LOBSTER
Cluster: CLAIX
Publications
Wuttig, M., Schön, C.-F., Lötfering, J., Golub, P., Gatti, C. and Raty, J.-Y. (2023), Revisiting the Nature of Chemical Bonding in Chalcogenides to Explain and Design their Properties. Adv. Mater.. Accepted Author Manuscript 2208485. https://doi.org/10.1002/adma.202208485
C.-F. Schön, S. van Bergerem, C. Mattes, A. Yadav, M. Grohe, L. Kobbelt, M. Wuttig, Science advances 2022, 8, eade0828.