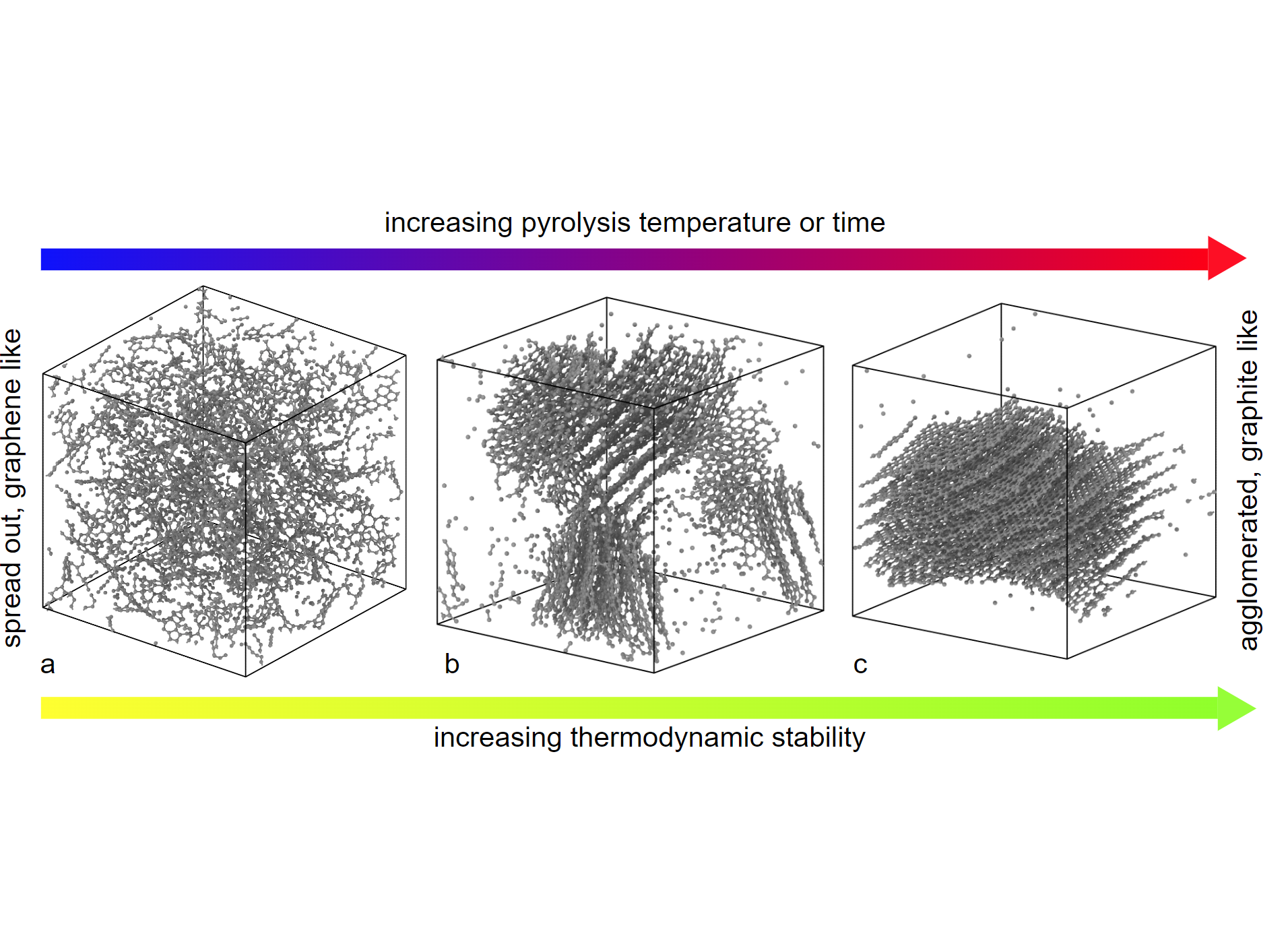
 Figure 1: Evolution of 'free carbon' phase in silicon oxycarbide glass-ceramics.
Figure 1: Evolution of 'free carbon' phase in silicon oxycarbide glass-ceramics. The simulation of materials on the atomistic scale requires a description of interatomic interactions. Quantum mechanical ab initio methods such as density functional theory (DFT) provide a high accuracy, but do not scale beyond system size of a few thousand atoms. Classical interatomic potentials (CIPs), on the other hand, use physically justified functional forms with few adjustable parameters to describe the interactions. They are suitable for simulations containing up to billions of atoms, but lack accuracy. Machine learning interatomic potentials (MLIPs) bridge the accuracy-scalability gap between DFT and CIPs. They employ flexible descriptor functions for atomic environments and machine learning techniques, allowing near DFT accuracy at a fraction of the computational cost. However, fitting them, i.e. adjusting their hundreds to thousands of coefficients, requires large amounts of training data. This training data is produced with DFT calculations. In this project training data sets were produced and corresponding MLIPs were fitted for Al-Cu-Zr, Mo-Si-O-C, and layered transition metal carbides.
Project Details
Project term
October 1, 2022–December 31, 2023
Affiliations
TU Darmstadt
Institute
Materials Modeling Division
Project Manager
Principal Investigator
Methods
MLIPs are a very active field of research and a variety of different types exist. In this
project mainly the atomic cluster expansion (ACE) formalism, which is a computationally
very efficient scheme, was employed. The training data was produced using plane-wave DFT as implemented in VASP and GPAW. In order to obtain training data covering a wide configurational space an iterative approach consisting of the following steps was employed:
1. Create initial training data based on simple structures and public repositories.
2. Calculate training energies and forces with DFT.
3. Train a MLIP.
4. Employ the MLIP in MD simulations to find unknown structures. Convergence is
reached when no new structures are found.
5. Add structures to the training data and repeat from step 3.
Developed potentials were employed in molecular dynamics (MD) simulations using
LAMMPS.
Results
A previously developed Cu-Zr database was extended with Al and different MLIPs were fitted to the training data. These MLIPs are currently evaluated with respect to their accuracy, speed and necessary amount of training data. Preliminary results suggest that the ACE framework employed for the other materials in this project offers a very good compromise between achievable accuracy and computational cost. MLIPs integrating modern message passing techniques achieve slightly better accuracies, but are around two orders of magnitude slower, both on CPUs and on GPUs. In the case of layered transition metal carbides a training database for Ti-Al-C in an aqueous HF solution (H, F and O) was generated. The resulting ACE potential is currently not able to model the aqueous solution with the desired accuracy. Consequently, it is undergoing further refinement. Here different strategies are employed. The simulation of water in DFT is difficult and typically employed exchange-correlation functionals fail reproduce several of its properties, such as the density, leading to inconsistencies within the training data itself. Consequently, a reduction to the Ti-Al-C system itself and the usage of different exchange-correlations functionals are considered for further developments. For Mo-Si-O-C a Mo-Si and Si-O-C training data were produced. Corresponding ACE potentials were developed and employed to investigate the structure and mechanical properties of the systems. Both potentials show a good accuracy over the whole compositional range, including various crystalline and amorphous phases. In a next step, the datasets will be combined and eventually further extended for a full Mo-Si-O-C MLIP.
Discussion
In this project MLIPs were fitted to a variety of material systems with a high degree of chemical and structural complexity. These MLIPs achieve higher accuracies than comparable CIPs, or are the first fitted to corresponding systems. They can be employed in MD simulations to investigate structural, mechanical and thermodynamical properties. The diversity of systems and properties investigated show the enormous flexibility of the approach, which allows new insights into materials on the atomistic scale.
Additional Project Information
DFG classification: 406 Materials Science
Software: VASP, LAMMPS
Cluster: Lichtenberg
Publications
Leimeroth, N.; Rohrer, J.; Albe, K.: “Structure-Property Relations of Silicon Oxycarbides Studied Using a Machine Learning Interatomic Potential.”, 2024.
https://doi.org/10.1111/jace.19932
Niklas Leimeroth: “Atomistic simulation based investigation of structure-property relations in silicon oxycarbides”, NHR conference 2023, Berlin, Germany
Ali M. Malik: “Predicting Exfoliation of MXenes via Ab-initio Thermodynamics”, EUROMAT
2023, Frankfurt, Germany
Ali M. Malik: “Ab-initio High-Throughput Screening for Magnetic MAX phases”, EMRS
2023, Strasbourg, France
Jochen Rohrer: “Towards in-depth atomistic understanding of polymer-derived silicon
oxycarbides using machine-learning potentials”, DPG spring meeting 2023, Berlin, Germany