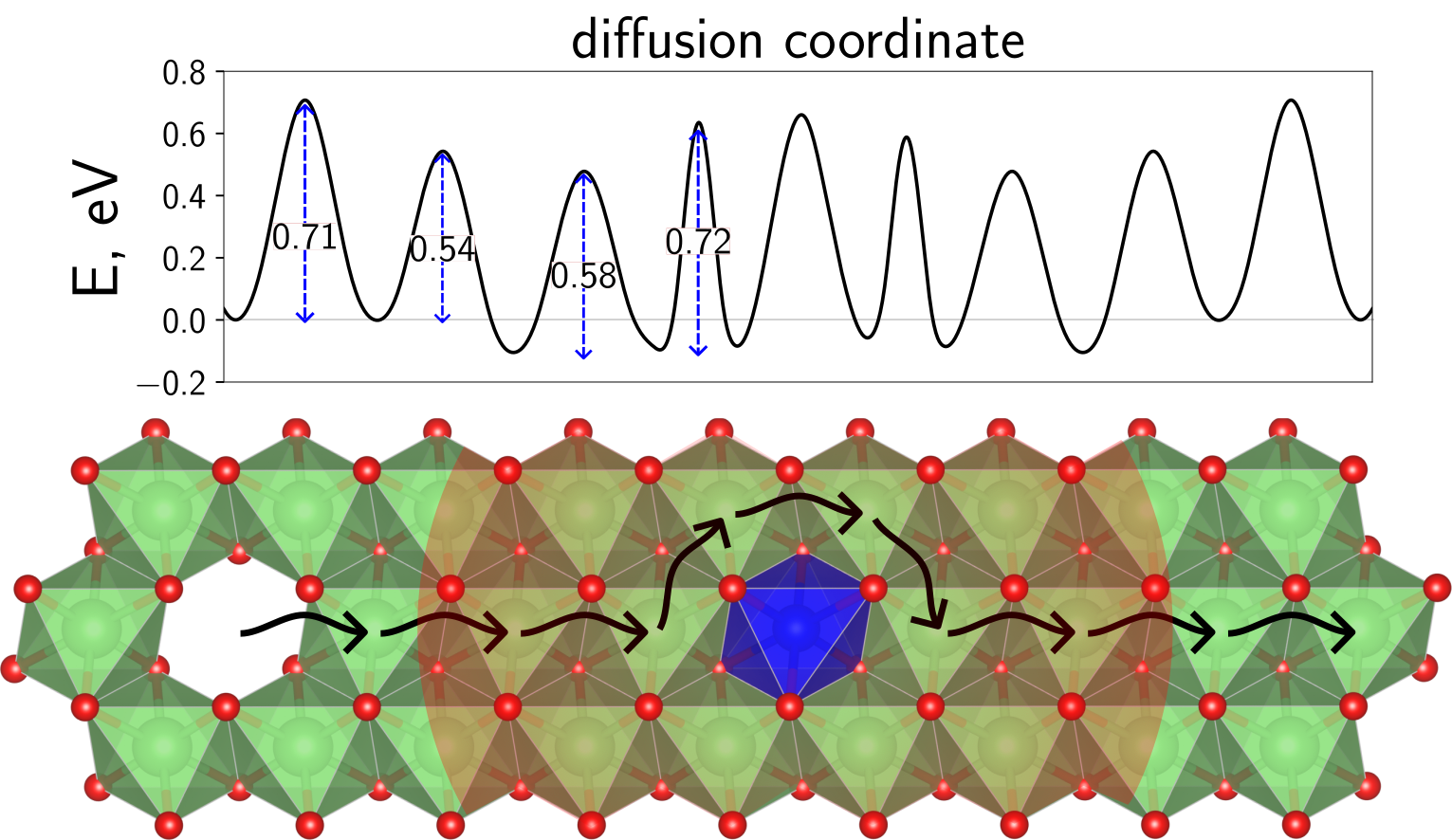
 Figure 1: Region of influence around NiLi defects in LiNiO2 at high Li content.
Figure 1: Region of influence around NiLi defects in LiNiO2 at high Li content. Layered transition metal oxides, derived from the model system LiCoO2, are used as cathode materials in Li-ion batteries. Currently, there are attempts to push to cathode materials to high Ni-contents, ultimately approaching LiNiO2. However, chemical and structural instabilities are observed for such materials upon cycling. By analyzing the chemomechanics of LiNiO2 with atomistic simulation methods, this projects aims to understand degradation mechanisms to help developing of improved cathode materials.
Project Details
Project term
February 1, 2023–January 31, 2024
Affiliations
TU Darmstadt
Institute
Materials Modeling Division
Project Manager
Principal Investigator
Methods
To be able to analyze the mechanisms of interest, atomistically resolved simulations techniques are necessary. To this end, we mainly focus on two approaches. First, we make use of density functional theory (DFT) simulations, which is the state-of-the-art method to computationally investigate the electronic structure in the course of material research. However, DFT is computationally costly and can only treat a limited system size. Therefore, we use the generated DFT data to train a machine learned interatomic potential to be able to analyze larger system sizes.
Results
The project was divided into four subprojects. Within the first subproject, we generated models of edge-dislocations. Such models need to be large in size and push the limits of DFT simulations. Therefore, this subproject took longer than expected and the results of these simulations are still analyzed. Within the second subproject, we originally planned to deal with the interactions of edge-dislocations and point defects such as Li and O vacancies. Unfortunately, due to the delay of the first subproject, the second subproject could not be completed. However, we used the provided computational time to investigate how native NiLi defects (generated during synthesis) are interacting with Li-ions and Li vacancies. These insights are not only important to understand Li diffusion in the bulk material, but also to compare the implications of point defects to dislocations at a later stage. A extensive database of LiNiO2-derived structures has been generated within the third subproject. This includes structures at different Li contents, rattled and distorted structures as well as structures derived from active learning cycles of molecular dynamics simulations at elevated temperatures. The database has been used to fit a machine learned interatomic potential based on the atomic cluster expansion (ACE) approach, which has been tested within the fourth subproject.
Discussion
Despite their importance for plastic deformations, the atomistic treatment of dislocations is hardly attempted in literature for complex materials that go beyond elemental metals or simple cubic systems. This is because anisotropic as well as electrostatic contributions from the presence of charged species heavily complicates the task of generating feasible starting structures, simulating the system under reasonable conditions and analyzing the results. Therefore, our investigations of dislocations in layered transition metal oxides mark a steppingstone, potentially inspiring further analyzes also in other research fields. Apart from dislocations, and more specifically to the properties of the investigated system LiNiO2, we could reveal how native NiLi defects change the potential energy landscape of approaching Li vacancies: Li vacancies are attracted by NiLi with a negative binding energy of roughly -0.1 eV. Moreover, NiLi lowers the barriers for approaching Li vacancies by almost 0.2 eV, possibly explaining the degraded kinetics observed during cycling towards high Li contents.
The generated ACE potential based on the described results and a targeted database shows high promise and will allow the treatment of larger systems and longer time scales. For example, even without the inclusion of dislocation structures, the potential has been able to run short molecular dynamics simulations under high shear with a screw dislocation present. Adding our dislocation models, refitting and performing more active learning cycles within a future project will help to generate a robust potential to analyze chemomechanical effects in LiNiO2.
Additional Project Information
DFG classification: 406 Materials Science
Software: VASP, LAMMPS
Cluster: Lichtenberg
Publications
Sicolo, S; Sadowski, M; Vettori, K; Bianchini, M; Janek, J; Albe, K. Off-Stoichiometry, Vacancy Trapping, and Pseudo-irreversible First-Cycle Capacity in LiNiO2, Chem. Mater. 2024, 36, 1, 492–500. https://doi.org/10.1021/acs.chemmater.3c02534
Sadowski, M; Sicolo, S; Albe, K. Planar Gliding and Vacancy Condensation: The role of Dislocations ain the Chemomechanical Degradation of Layered Transition-Metal Oxides, Lithium Battery Discussions “Electrode Materials”, 18.-23. Juni 2023, Arcachon, Frankreich